
Lacidipine synthesis
- Product Name:Lacidipine
- CAS Number:103890-78-4
- Molecular formula:C26H33NO6
- Molecular Weight:455.54
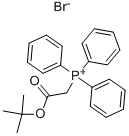
59159-39-6
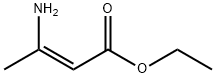
626-34-6

643-79-8
103890-78-4
1. 18 liters of dichloromethane were added to the reactor, followed by 5 kg of tert-butoxycarbonylmethyltriphenylphosphonium bromide (prepared with reference to Example 1). 2. 2.05 kg of o-phthalaldehyde was added, followed by 1 liter of dichloromethane, and the mixture was stirred for about 15 minutes, then cooled to -5°C. 3. A solution of 2.65 kg of sodium hydroxide flakes in 5 liters of water (pre-prepared at 25°C) was added to the reaction mixture at -3°C and kept at -3°C for 2.5 hours. The reaction process was monitored by thin layer chromatography. 4. After completion of the reaction, the mixture was warmed up to 25°C and stirring was continued for 30 minutes. The organic layer was separated and the solvent was removed by atmospheric pressure distillation at 52°C. The residue was kept at 52°C for 15 minutes. 5. 35 liters of n-heptane were added and 4 liters of n-heptane were distilled under 500 mmHg vacuum and at 63 °C. The mixture was then cooled to 0°C and held at 35°C for 1.5 hours. 6. The insoluble material was removed by vacuum filtration and the filter cake was washed with 7.5 liters of n-heptane. The filtrate was transferred to another reactor and the solvent was removed by complete distillation under vacuum at 610 mmHg and 68°C. 7. Cooled to 33 °C, 11.5 liters of isopropanol was added and subsequently cooled to -7 °C. 8. At -7 °C, a solution of 4.25 kg ethyl 3-aminocrotonate in 12.5 liters of isopropanol was slowly added, followed by 2.8 liters of trifluoroacetic acid and 1 liter of isopropanol, and the reaction was kept at -7 °C for 2 hours and 45 minutes. The reaction process was monitored by thin layer chromatography. 9. A solution of 2.6 kg of sodium bicarbonate in 50 liters of water was added to the reaction mixture at 0 °C, followed by 25 liters of ethyl acetate. Increase the temperature to 25°C and stir for 20 minutes. Separate the aqueous layer and extract with 12.5 liters of ethyl acetate. The organic layers were combined and the solvent was removed by atmospheric pressure distillation at 81 °C. 10. Add 5 liters of isopropanol to the residue and distill completely to remove the solvent. This operation is repeated once. Finally, the residue was dissolved in 63 liters of isopropanol and heated to 85°C to obtain a clarified solution. 11. Cool to 2 °C, keep for 1.5 h. Separate the solid by filtration and wash with 2.5 liters of isopropanol. The wet product was dried at 50°C for 30 minutes. 12. The dried substance was added to 23 liters of isopropanol, heated to 65°C and held for 30 minutes, cooled to 2°C and held for 1 hour, filtered and washed with 1.5 liters of isopropanol. 13. The wet solid was dried at 60 °C and 630 mmHg under vacuum for 5 h to give 2.04 kg of ethyl (E)-diethyl 4-(2-(3-(tert-butoxy)-3-oxoprop-1-en-1-yl)phenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate in 40.9% yield. The content of triphenylphosphine oxide was less than 0.0011 area%.
59159-39-6
108 suppliers
$8.00/1g
626-34-6
218 suppliers
$6.00/5g
643-79-8
733 suppliers
$9.00/1g
103890-78-4
319 suppliers
$6.00/1g
Yield:103890-78-4 40.9%
Reaction Conditions:
Stage #1:(tert-butoxycarbonylmethyl)triphenylphosphonium bromide;o-phthalic dicarboxaldehyde with sodium hydroxide in dichloromethane;water at -5 - -3; for 2.75 h;Industry scale;
Stage #2:ethyl 3-aminobut-2-enoate with trifluoroacetic acid in isopropyl alcohol at -7; for 2.75 h;
Stage #3: with sodium hydrogencarbonate in water;ethyl acetate;isopropyl alcohol at 0 - 25; for 0.333333 h;
Steps:
2
18 liters of dichloromethane was taken into a reactor and 5 kg of tertiary-butoxy carbonyl methyl triphenyl phosphonium bromide (obtained using a similar process as described in Example 1) was added to it. 2.05 kg of ortho-phtalaldehyde was added to the reaction mass and another 1 liter of dichloromethane was added to it. The reaction mass was stirred for about 15 minutes and then cooled to about -5° C. A solution of 2.65 kg of sodium hydroxide flakes in 5 liters of water at about 25° C. was added to the reaction mass at -3° C. and maintained at -3° C. for 2.5 hours. Reaction completion was checked using thin layer chromatography. After the reaction was completed, the temperature of the reaction mass was raised to 25° C. and stirred for 30 minutes. The organic layer was separated from the reaction mass and distilled without vacuum at a temperature of 52° C. The residue obtained was maintained at 52° C. for 15 minutes. 35 liters of n-heptane was added to the reaction mass at 52° C. 4 liters of the n-heptane was distilled off from the reaction mass under a vacuum of 500 mm Hg at 63° C. The reaction mass was then cooled to 35° C. and maintained for 1.5 hours. The reaction mass was then filtered under vacuum to remove the undissolved material. The filtered cake was washed with 7.5 liters of n-heptane. The filtrate was taken into another reactor and the solvent was distilled off completely under a vacuum of 610 mm Hg and at 68° C. The reaction mass was then cooled to 33° C. and 11.5 liters of isopropanol was charged into it. The reaction mass was then cooled to -7° C. A solution of 4.25 kg of ethyl-3-aminocrotonate in 12.5 liters of isopropanol was added to the reaction mass at -7° C. 2.8 liters of trifluoroacetic acid was added to the reaction mass at -7° C. followed by addition of 1 liter of isopropanol. The reaction mass was maintained at -7° C. for 2 hours 45 minutes. Reaction completion was checked using thin layer chromatography. A solution of 2.6 kg of sodium bicarbonate in 50 liters of water was added to the reaction mass 0° C. and 25 liters of ethyl acetate was added to it. The temperature of the reaction mass was heated to 25° C. and stirred for 20 minutes. The aqueous layer was separated and extracted with 12.5 liters of ethyl acetate. The combined organic layer was taken into a separate reactor and the solvent was distilled off atmospherically at 81° C. The residue was distilled off completely, and then 5 liters of isopropanol was added to it. The reaction mass was distilled off completely and again 5 liters of isopropanol was added to the residue obtained. The reaction mass was again distilled off completely and finally the residue was dissolved in 63 liters of isopropanol by heating to 85° C. to get clear dissolution. The reaction mass was then cooled to 2° C. and maintained for 1.5 hours. The isolated material was filtered and washed with 2.5 liters of isopropanol. The wet material was dried at 50° C. for 30 minutes. The dry material was taken into another reactor and 23 liters of isopropanol was added to it. The reaction mass was heated to 65° C. and maintained for 30 minutes. The reaction mass was then cooled to 2° C. and maintained for 1 hour. The reaction mass was filtered and washed with 1.5 liters of isopropanol. The wet solid was dried at a temperature of 60° C. and a vacuum of 630 mm Hg for 5 hours to yield 2.04 kg (yield: 40.9%) of the title compound. Triphenyl phosphine oxide content: less than 0.0011 area-%.
References:
Sajja, Eswaraiah;Vetukuri, Venkata Naga Kali Vara Prasada Raju;Ningam, Srinivas Reddy;Vedantham, Ravindra US2007/43088, 2007, A1 Location in patent:Page/Page column 6
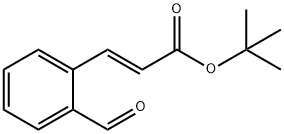
103890-69-3
52 suppliers
inquiry

144-55-8
1392 suppliers
$10.00/25g
626-34-6
218 suppliers
$6.00/5g
103890-78-4
319 suppliers
$6.00/1g