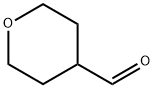
Tetrahydropyran-4-carbaldehyde synthesis
- Product Name:Tetrahydropyran-4-carbaldehyde
- CAS Number:50675-18-8
- Molecular formula:C6H10O2
- Molecular Weight:114.14

4295-99-2
50675-18-8
General procedure for the synthesis of tetrahydropyran-4-carbaldehyde from 4-cyanotetrahydropyran: Referring to Example 2. To a solution of toluene (10 mL) containing tetrahydropyran-4-carbonitrile (1.0 g, 9.0 mmol) was slowly added, at -78° C., a solution of diisobutylaluminum hydride (DIBAL-H, 10.8 mL, 10.8 mmol, 1.0 M of the toluene solution ). The reaction mixture was stirred continuously at -78 °C for 1 h, followed by slow warming to room temperature. Upon completion of the reaction, the reaction was quenched with saturated ammonium chloride solution and extracted with ethyl acetate. The organic phases were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the target product tetrahydropyran-4-carbaldehyde (530 mg, 52% yield).
4295-99-2
168 suppliers
inquiry
50675-18-8
217 suppliers
$15.00/100mg
Yield:50675-18-8 80%
Reaction Conditions:
with diisobutylaluminium hydride in toluene at -5 - 25;Inert atmosphere;
Steps:
Tetrahydropyran-4-carbaldehyde (6)
[0169] Dry toluene (500 g) was charged into a dried reactor under nitrogen and tetrahydropyran-4-carbonitrile 5 (100 g, 0.90 mol) was added. The resulting solution was cooled to -5°C to 5°C and a solution of DIBAL-H in toluene (1.0M, 800 g, 1.0 mol) was added dropwise at -5°C to 5°C and the resulting reaction mxture was stirred at this temperature for 1-2 hrs. The reaction mixture was then warmed to 20-25°C and stirred at this temperature for 1-2 hrs. Subsequently the reaction mixture was cooled to -5°C - 5°C and quenched by the dropwise addition of a solution of AcOH (195 g) in toluene (180 g) at -5° - 5°C (caution: exothermic, gas release). A 25% solution of sodium tartrate tetrahydrate (1000 g) was added slowly at -5°C - 5°C (caution:exothermic, gas release). The reaction mixture was allowed to warm to 20-25°C and stirred at this temperature for 8-16 hrs. The layers were separated and the aqueous layer was extracted twice with EtOAc (900 mL each) at 20-25°C. The two EtOAc extracts were combined with the original organic phase and the combined organic phases were evaporated in vacuo to about 100-200 g to yield the product 6. The product was generally isolated in 50-80% yield.
References:
IMARA, INC.;SVENSTRUP, Niels;ZHANG, Jun;SUN, Jikui;CHEN, Yuyin;KONG, Jianshe;MA, Rujian;ZHANG, Junhua;QIN, Liang;XIAO, Huanming;SUN, Jinxu;MENG, Xiao;SUN, Fenglai;ZHU, Jingyang WO2018/218104, 2018, A1 Location in patent:Paragraph 0154; 0169
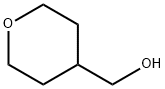
14774-37-9
256 suppliers
$5.00/1g
50675-18-8
217 suppliers
$15.00/100mg

101765-10-0
6 suppliers
inquiry
50675-18-8
217 suppliers
$15.00/100mg

110238-91-0
216 suppliers
$13.00/5g
50675-18-8
217 suppliers
$15.00/100mg
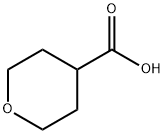
5337-03-1
311 suppliers
$10.00/1 g
50675-18-8
217 suppliers
$15.00/100mg